




















![Der Geist des digitalen Kapitalismus im Gesundheitswesen [Gesundheits-Check]](https://s0.wp.com/i/blank.jpg)
,regionOfInterest=(1626,2221)&hash=4cd2016f3da107f5274fad3de5da2cf5cbd13cbff2f89a3652ed30e38a3a086d#)









![Deals: Perfekte Videos - Diese Action-Cam macht eure GoPro überflüssig – für kurze Zeit günstiger! [Anzeige]](https://images.cgames.de/images/gamestar/4/dji-osmo-action-4_6354461.jpg?#)





























Medizinischer Erfolg: "Ganz sicher ein Durchbruch": Individualisierte Gentherapie wirkt bei schwerkrankem Baby
Ein Baby mit einer sehr seltenen, schweren Erbkrankheit ist erstmals erfolgreich mit einer personalisierten Gentherapie behandelt worden. Sollte die Technik langfristig helfen, könnten Millionen von Menschen davon profitieren
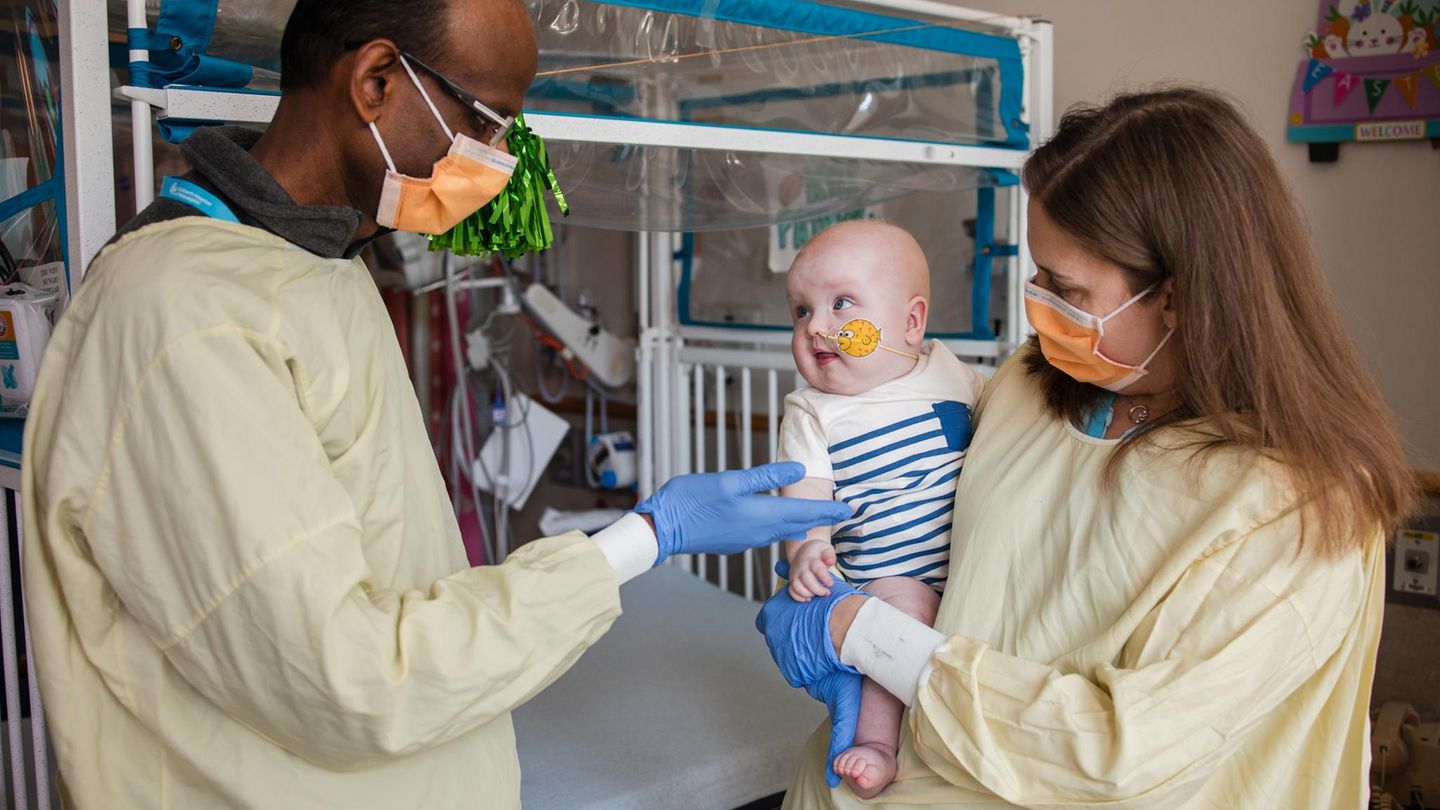
Ein Baby mit einer sehr seltenen, schweren Erbkrankheit ist erstmals erfolgreich mit einer personalisierten Gentherapie behandelt worden. Sollte die Technik langfristig helfen, könnten Millionen von Menschen davon profitieren
Erstmals hat eine individualisierte Gentherapie einem Baby mit einer schweren, seltenen Erbkrankheit geholfen. Das Baby KJ wurde in den USA mit einer lebensbedrohlichen Stoffwechselstörung geboren. Der Junge bekam im Februar 2025 – etwa sieben Monate nach der Geburt – seine erste Gentherapie, weitere folgten.
"Die Behandlung verlief sicher, und er wächst und gedeiht jetzt gut", schreibt das Children’s Hospital of Philadelphia, wo KJ behandelt wurde. Die ersten Monate habe er dagegen unter anderem mit einer extrem eingeschränkten Ernährung in der Klinik verbracht und symptomlindernde Medikamente erhalten. Sein Gendefekt führt zu einer Ansammlung von Ammoniak im Blut, was Nerven- und Hirnschäden verursachen kann.
Schon normale Kinderkrankheiten durchgestanden
Das Baby bekam laut Mitteilung der Klinik zwei weitere Gentherapie-Dosen. "Bis April 2025 hatte KJ drei Dosen erhalten – ohne ernsthafte Nebenwirkungen." In der kurzen Zeit seit der Therapie vertrage das Baby mehr Eiweiß in der Nahrung und benötige weniger Medikamente. Zudem habe es normale Kinderkrankheiten wie eine Erkältung gut überstanden.
Baby KJ kann endlich bei Eltern und Geschwistern sein
Seit KJs Geburt "dreht sich unsere ganze Welt um diesen kleinen Kerl und seinen Aufenthalt im Krankenhaus", wird Vater Kyle Muldoon in einer Mitteilung der Klinik zitiert. "Wir sind so froh, dass wir endlich zu Hause sein können, damit KJ bei seinen Geschwistern sein kann und wir endlich aufatmen können."
"Als die Ärzte mit ihrer Idee zu uns kamen, haben wir ihnen vertraut – in der Hoffnung, dass sie nicht nur KJ, sondern auch anderen Familien, die in derselben Situation sind wie wir, helfen könnte", sagt Mutter Nicole Muldoon.
Nachdem der Neugeborene die Diagnose eines schweren Mangels des Enzyms Carbamoylphosphat-Synthetase-1 erhalten hatte – einer Krankheit mit einer geschätzten Sterblichkeitsrate von 50 Prozent im frühen Säuglingsalter –, hat das Ärzteteam nach eigenen Angaben mit der Entwicklung der maßgeschneiderten Gentherapie begonnen. Dabei wird letztlich ein spezielles Werkzeug in Lipidtröpfchen verpackt und per Infusion in die Leber gegeben, wo es das Erbgut von vielen Zellen reparierte.
"Ganz sicher ein Durchbruch"
Mit dieser Technik könnte einmal Millionen Menschen geholfen werden, die an seltenen Erbkrankheiten leiden, sagt Arndt Borkhardt, Direktor der Klinik für Kinder-Onkologie, -Hämatologie und klinische Immunologie am Universitätsklinikum Düsseldorf. "Das ist ganz sicher ein Durchbruch."
Das Team präsentiert die Gentherapie im "New England Journal of Medicine" und auf dem Jahreskongress der American Society of Gene & Cell Therapy in New Orleans. "KJ ist zwar nur ein einzelner Patient, aber wir hoffen, dass er der erste von vielen ist, die von einer Methode profitieren, die individuell auf die Bedürfnisse von Patienten abgestimmt werden kann", sagt Mitautorin Rebecca Ahrens-Nicklas, Direktorin des Programms für Gentherapie bei vererbten Stoffwechselkrankheiten an der Klinik in Philadelphia.
Ein Erfolg, aber keine Heilung
Es seien allerdings noch längere Nachbeobachtungen erforderlich, um Sicherheit und Wirksamkeit der Therapie zu beurteilen, räumt das Forschungsteam ein. Prinzipiell könnte der Patient in Zukunft weitere und höhere Dosen erhalten, falls nötig. Bis die Therapie einmal Routine werden könnte, werden nach Expertenangaben noch Jahre vergehen.
Julian Grünewald von der Technischen Universität München verweist darauf, dass das Baby nach der Gentherapie weniger Medikamente benötigte und weniger Symptome hatte, aber noch keine "funktionelle Heilung" erfolgt sei. "Man muss aber auch bedenken, dass in einer solchen erstmaligen Testung die Sicherheitsaspekte im Vordergrund stehen. Daher wurden auch vergleichsweise geringe Dosierungen genutzt."
"Eine weitere Besserung und vollständige Heilung ist denkbar, falls man die Dosis sicher steigern kann", sagt Grünewald. "Alles in allem sind die Ergebnisse spektakulär für das Feld der Gen- und Zelltherapie und geben Hoffnung für die Behandlung seltener Erkrankungen."
Therapie baut auf Genschere von Charpentier und Doudna auf
Der Düsseldorfer Experte Borkhardt sieht zwei Besonderheiten der Studie: Die erste sei, dass es sich hier um eine richtige Korrektur des genetischen Defektes handle, ein sogenanntes Base Editing. Dabei werde ein einziger krank machender genetischer Baustein (Base) gegen einen anderen Baustein ausgetauscht. "Bisherige Crispr-Gentherapien schneiden DNA-Abschnitte, also zerstören die DNA und setzen sie wieder zusammen." Das sei bei den zugelassenen Gentherapien gegen die Blutbildungsstörungen Thalassämie und Sichelzellkrankheit der Fall. Dabei könnten mehr Fehler passieren, sodass sie wesentlich risikoreicher seien.
"Aber die zweite Besonderheit dieser Arbeit ist wahrscheinlich noch sehr viel weitreichender. Das ist die Individualisierung der Gentherapie", sagt Borkhardt. Bei den beiden eben genannten Erkrankungen würden mit der Genschere Crispr/Cas jeweils viele Menschen therapiert, die denselben Gendefekt haben – dabei wird jeweils ein Gen ausgeschaltet.
Die aktuelle Studie baut zwar ebenfalls auf der von den Nobelpreisträgerinnen Emmanuelle Charpentier und Jennifer Doudna entwickelten Genschere Crispr/Cas auf. Das Team hat aber eine weiterentwickelte Technik genutzt.
Individuelle Gentherapie wurde in sehr kurzer Zeit entwickelt
"Hier ist es so, dass man erstmals die Möglichkeit hat, in das Gebiet der seltenen oder ultraseltenen Erkrankungen zu gelangen, wo der Gendefekt sehr individuell ist", sagt Borkhardt. Ein Gen könne an vielen Stellen defekt sein und jeder Patient habe seine eigene Mutation.
Derzeit könne man bereits innerhalb von Tagen oder einer Woche nach der Geburt eine genetische Krankheit feststellen, so Borkhardt. "Was natürlich jetzt hier völlig neu ist, dass man eine Gentherapie designt in sehr kurzer Zeit." KJ habe ein krank machendes Gen (Allel) für ein Enzym vom Vater und eines von der Mutter erhalten.
"Das nächste Baby mit derselben Erkrankung hat voraussichtlich ganz andere Mutationen und bräuchte natürlich dann ein ganz anderes gentherapeutisches Produkt", erläutert Borkhardt. Dann müssten ganz andere Genbausteine korrigiert werden. "Das Team hat jetzt jedoch eine Pipeline aufgebaut, dass man in kurzer Zeit, unabhängig von der individuell vorliegenden Mutation, eine Korrektur durchführen kann."
Wie das Werkzeug funktioniert
Und das geht so: Das Werkzeug besteht zum einen aus einer sogenannten guide RNA (gRNA), die den defekten Genbaustein findet. Diese lässt sich leicht an jedes Erbgut anpassen. Zum anderen enthält es ein Protein, das die Gen-Bausteine austauscht. Verpackt ist es in einem Lipidtröpfchen.
Theoretisch könne die Therapie auch für ältere Kinder und Erwachsene genutzt werden, sagt Borkhardt. Er warnt jedoch vor zu großen Hoffnungen auf rasche Hilfe. Die Therapie sei komplex. Für die klinische Routine werde es Jahre dauern, bis solche Therapiemöglichkeiten flächendeckend aufgebaut seien.
Bei der Studie handle es sich um eine großartige Demonstration, "aber es sei auch darauf hingewiesen, dass die Korrektur in der Leber erfolgte", sagt Marc Güell von der Universität Pompeu Fabra in Barcelona. Andere Gewebe ließen sich derzeit noch wesentlich schwerer gentherapeutisch behandeln.
Der hier verfolgte Prozess sei zudem äußerst komplex, und es werde viel Arbeit nötig sein, um herauszufinden, wie man ihn weiter verbreiten und auf andere Fälle ausweiten könne. Borkhardt hält am ehesten eine Ausweitung der Therapie für Erkrankungen mit individueller Mutation im Knochenmark für möglich.
Längere Beobachtung nötig
Über die Risiken lasse sich derzeit noch wenig sagen, meint Borkhardt. "Das braucht natürlich, das sagen die Autoren selbst, eine viel längere Nachbeobachtung." Das Kind müsse sicher weiterhin nachbetreut werden, um zu schauen: "Ist es stabil? Bleibt diese Genkorrektur so, dass auch die Stoffwechselleistung der Leber ausreicht, lebenslang ihren Dienst zu tun? Das kann man heute natürlich alles nicht sagen mit diesem kurzen Follow-up."
Für Lluís Montoliu vom spanischen Centro Nacional de Biotecnología in Madrid sind die entscheidenden offenen Fragen: "Wie teuer wird eine solche Therapie sein? Wo kann sie durchgeführt werden? Wie viele Kinder werden von dieser neuen individualisierten Therapie profitieren?"
In Deutschland geschätzt vier Millionen Menschen betroffen
In der EU gilt eine Erkrankung dann als selten, wenn sie höchstens 5 von 10.000 Menschen haben. Derzeit sind laut Robert Koch-Institut etwa 8.000 Seltene Erkrankungen bekannt. Sie sind demnach überwiegend genetisch bedingt und haben oft schwere, chronische Verläufe. In Deutschland seien geschätzt vier Millionen Menschen davon betroffen.
"Wir hoffen, dass andere akademische Forscher diese Methode für viele seltene Krankheiten reproduzieren und vielen Patienten eine faire Chance auf ein gesundes Leben geben", sagt Erstautor Kiran Musunuru von der behandelnden Kinderklinik in Philadelphia.


